



 Oligo Pools
Oligo Pools
 Variant Libraries
Variant Libraries








Twist EM-seq v2 Methylation Detection System
- Overview
- Resources
- Application
Twist EM-seq v2 Methylation Detection System enables accurate methylation analysis through a fully enzymatic workflow designed to preserve DNA integrity while delivering consistent library yields and high conversion efficiency. Optimized for low input and challenging samples, EM-Seq v2 supports robust methylation profiling without bisulfite induced DNA damage.


 Product Sheet
Product Sheet
 Protocol
Protocol
Product data
Engineered reagents preserve performance post-conversion
Libraries prepared with EM-Seq v2 maintain capture efficiency and uniformity. Performance is preserved through downstream workflows specifically for methylation applications, including compatibility with both standard and fast hybridization protocols.
Consistent Methylation Hybridization Capture Performance
EM-Seq v2 libraries processed with Twist hybridization maintain consistent capture efficiency and methylation performance, supporting reliable downstream analysis.
Reproducible Methylation Profiling Across Targets
Consistent detection of differentially methylated regions demonstrates reliable performance across methylation targets, reinforcing reproducibility for downstream methylation analysis.
Consistent library yield across DNA inputs
EM-Seq v2 generates consistent, usable library yields across a wide DNA input range. Reduced dropout at lower inputs supports methylation sequencing when sample availability is limited, while maintaining reliable performance across workflows.
Confident Enzymatic Conversion for Accurate CpG Methylation Calling
EM-Seq v2 delivers robust enzymatic conversion of unmethylated cytosines with consistent CpG call ratios, supporting accurate and reliable methylation analysis while preserving DNA integrity.
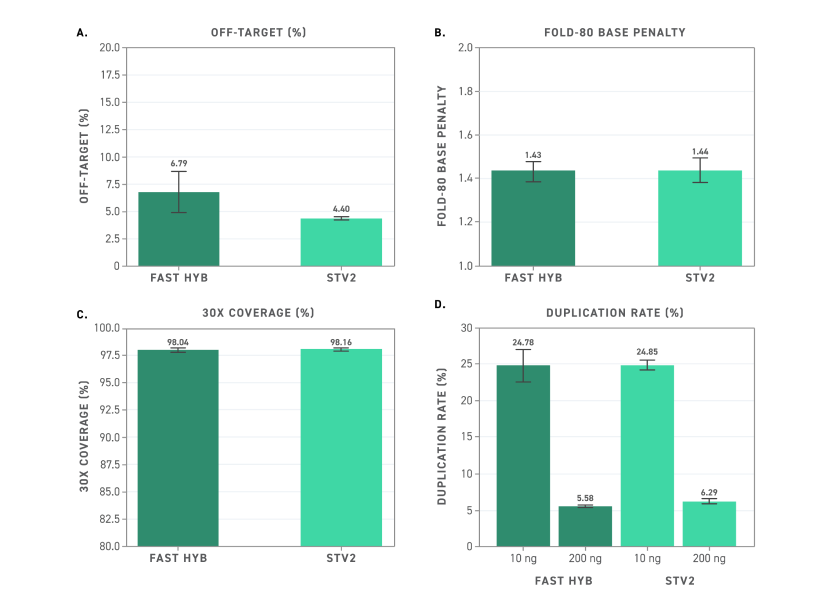
Figure 1. The Twist Methylation Enhancer improves target enrichment quality and reduces methylation-specific off-target hybridization for Twist EM-Seq v2 libraries captured with the Twist Alliance Pan-Cancer panel, using either the Fast or Standard Hybridization v2 systems.
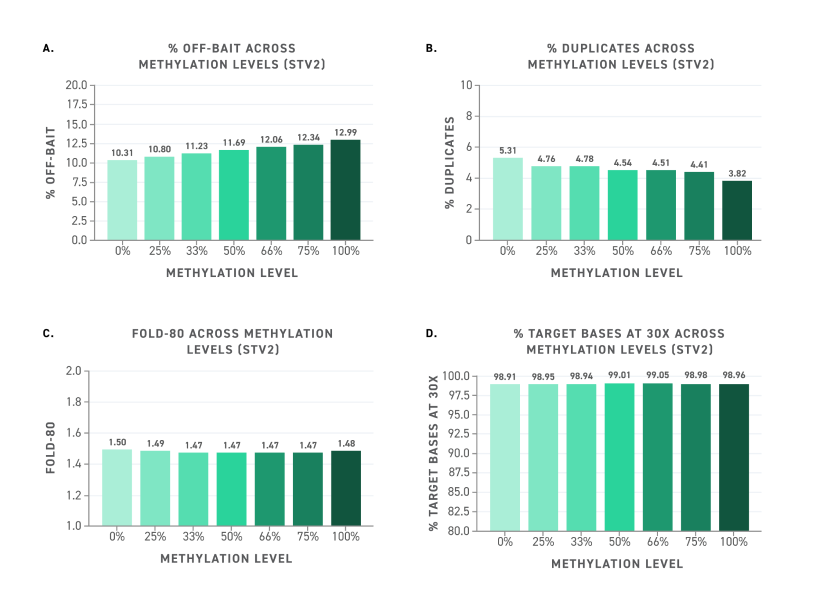
Figure 2. Twist EM-Seq v2 Methylation Detection System libraries captured with the Twist Alliance Pan-Caner panel using the Twist Standard Hybridization v2 system provide highly reproducible Picard target enrichment performance metrics across samples with highly variable methylation levels.

Figure 3. Highly sensitive methylation detection. Detection of methylation is possible across a wide range of methylation levels and targets.
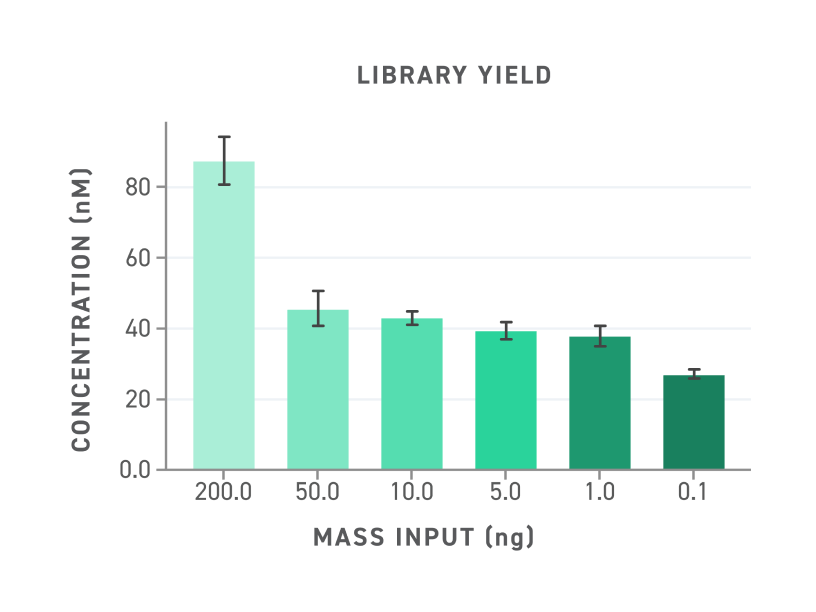
Figure 4. Yield of libraries generated with Twist EM-seq v2 Methylation Detection System. Library yields are high even with low mass input.
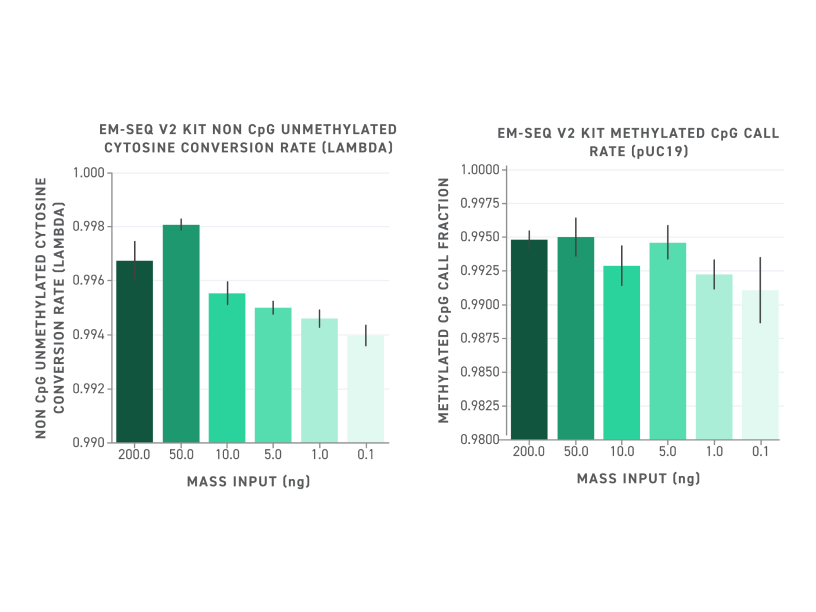
Figure 5. Left Panel: Non-CPG Conversion Rate. Conversion rates with v2 are high even with low mass input. Right Panel: Called CPG Ratio. Conversion specificity with v2 is high even with low mass input.
Engineered reagents preserve performance post-conversion
Libraries prepared with EM-Seq v2 maintain capture efficiency and uniformity. Performance is preserved through downstream workflows specifically for methylation applications, including compatibility with both standard and fast hybridization protocols.
Figure 1. The Twist Methylation Enhancer improves target enrichment quality and reduces methylation-specific off-target hybridization for Twist EM-Seq v2 libraries captured with the Twist Alliance Pan-Cancer panel, using either the Fast or Standard Hybridization v2 systems.
Consistent Methylation Hybridization Capture Performance
EM-Seq v2 libraries processed with Twist hybridization maintain consistent capture efficiency and methylation performance, supporting reliable downstream analysis.
Figure 2. Twist EM-Seq v2 Methylation Detection System libraries captured with the Twist Alliance Pan-Caner panel using the Twist Standard Hybridization v2 system provide highly reproducible Picard target enrichment performance metrics across samples with highly variable methylation levels.
Reproducible Methylation Profiling Across Targets
Consistent detection of differentially methylated regions demonstrates reliable performance across methylation targets, reinforcing reproducibility for downstream methylation analysis.

Figure 3. Highly sensitive methylation detection. Detection of methylation is possible across a wide range of methylation levels and targets.
Consistent library yield across DNA inputs
EM-Seq v2 generates consistent, usable library yields across a wide DNA input range. Reduced dropout at lower inputs supports methylation sequencing when sample availability is limited, while maintaining reliable performance across workflows.
Figure 4. Yield of libraries generated with Twist EM-seq v2 Methylation Detection System. Library yields are high even with low mass input.
Confident Enzymatic Conversion for Accurate CpG Methylation Calling
EM-Seq v2 delivers robust enzymatic conversion of unmethylated cytosines with consistent CpG call ratios, supporting accurate and reliable methylation analysis while preserving DNA integrity.
Figure 5. Left Panel: Non-CPG Conversion Rate. Conversion rates with v2 are high even with low mass input. Right Panel: Called CPG Ratio. Conversion specificity with v2 is high even with low mass input.
Product Data
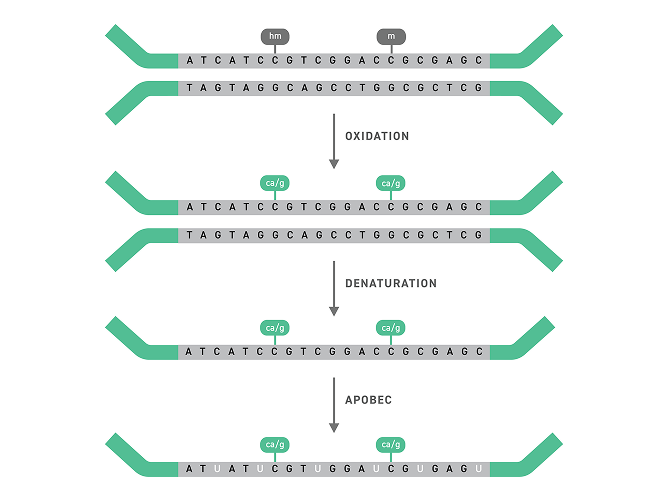
Enzymatic Methylation Conversion with EM-Seq v2
EM-Seq V2 uses enzymatic reactions to selectively convert unmethylated cytosines while protecting methylated bases. After amplification, unmethylated sites are read as thymine and methylated sites remain cytosine, enabling accurate methylation analysis without DNA damage.
Highlighted resources

 Product Sheet
Product SheetRelated Workflows and Applications
Target Enrichment Workflow
Target enrichment workflows with Twist deliver scalable, low bias, and highly uniform coverage across large and complex genomic regions; enabling broader discovery, higher sensitivity, and greater flexibility than amplicon or array-based methods.
Library Preparation Solutions
Library PrepTwist Library Preparation Kits streamline the construction of high-quality DNA libraries for next-generation sequencing by combining key preparation steps into a single, efficient reaction.
Learn More 
Custom Panels
Target Enrichment PanelsTwist Custom NGS Panels let you move beyond off-the-shelf solutions, giving you full control over what you sequence.
Learn More
Liquid Biopsy application
Twist offers several tools to support research into liquid biopsies and early cancer characterization.
Library Preparation Solutions
Library PrepTwist Library Preparation Kits streamline the construction of high-quality DNA libraries for next-generation sequencing by combining key preparation steps into a single, efficient reaction.
Learn More
Oncology - DNA CGP Panel
Target Enrichment PanelsThe Twist DNA CGP Panel enables comprehensive detection of SNVs, indels, copy number alterations, and gene rearrangements, delivering broad mutation coverage in a single assay.
Learn More
MRD Rapid 500 Panel
Target Enrichment PanelsThe Twist Bioscience MRD Rapid 500 Panel is a scalable target enrichment solution for minimal residual disease, enabling broad genomic coverage with panels ranging from 50 to 500 probes.
Learn More

Twist Bioscience HQ
681 Gateway Blvd
South San Francisco, CA 94080