



 Oligo Pools
Oligo Pools
 Variant Libraries
Variant Libraries








Get the latest by subscribing to our blog
As we often discuss here on our blog, protein engineering is a complex task that pits researchers against nature. Natural proteins are evolved for their function in their native cellular setting, and often don’t perform optimally when they are co-opted for chemical engineering. As a result, biochemists require proteins that have been engineered for optimal properties in a non-natural setting.
Recent independent research by Professor Manfred Reetz has shown that Twist Bioscience’s variant libraries are powerful, near bias-free tools for enzyme engineering. In a new paper, Professor Reetz’s lab reinforces this finding, showing that in research using synthesised libraries for protein engineering, Twist Bioscience’s libraries had the highest ratings. We take a detailed look at this new research here.
Last year, Professor Manfred Reetz and colleagues at the Max Planck institute in Mulheim, Germany, published a study exploring the use of Twist Bioscience’s variant libraries for the engineering of better biocatalysts. While many enzyme engineers rely on randomization methods, like degenerate primer PCR, Professor Reetz focused on the direct solid-phase synthesis of variant libraries for enzyme engineering from Twist Bioscience. The study, conducted independently from Twist Bioscience, focused on biases introduced by different methods of library construction into enzyme libraries, as an assessment of library quality at the DNA level.
In an interview with Professor Reetz published on our blog, he described how traditional library generation methods introduce bias toward certain sequences. Such biases mean that some sequences are significantly underrepresented in the the entire library, and considerable screening efforts are required to capture all of the variation that is designed in the experiment. This means useful variants could be missed, and experiments can have an increased failure rate when it comes to generating useful enzymes. On the other hand, Professor Reetz’s research uncovered that variant libraries generated by solid phase DNA synthesis by Twist Bioscience contained exceptionally low bias, and are likely to be an essential tool in the future of protein engineering.

Professor Manfred Reetz (right) with the lead author on his laboratory’s previous Twist library mediated protein engineering paper, Dr. Aitao Li.
After three rounds of screening a Twist Bioscience variant library, 97% of the designed variation was obtained. Massive sequencing of the library showed that an almost perfectly even distribution of variants was also present. In an equivalent PCR-based library, only 54% of the variation was obtained in the same screening effort, and their distribution of designed variation was very uneven. Additionally, double the amount of useful, enantioselective protein variants were isolated from the Twist Bioscience library compared to the PCR-based library, showing that the direct synthesis of protein variants is a powerful tool for maximising engineering output within a typical design-build-test cycle.
In continuation of this work, Professor Reetz’s lab recently published a new report titled “Solid-Phase Gene Synthesis for Mutant Library Construction: The Future of Directed Evolution?” This work reviews a protein engineer’s options when it comes to removing bias in high throughput engineering pipelines and reviews the solid-phase DNA synthesis libraries available with Twist Bioscience receiving the highest rating.
High throughput protein engineering involves the generation of a library of amino acid variants of the protein that is then screened for improved properties - a process termed “directed evolution.” However, the design of such libraries is no simple task. Protein sequence space is enormous. A small protein of 100 amino acids contains 1040 of possible variations, which is 16 orders of magnitude more variation than there are stars in the observable universe.
Scientists have developed tools that circumvent this numbers problem, selecting optimal amino acids in which to introduce variation and the optimal set of variants within those residues. For example Professor Reetz’s lab developed a workflow called CASTing (Combinatorial Active-site Saturation Test) that hones in on only amino acids in the active site that give the best possible chance of modifying the enzyme’s catalytic function. By randomizing these amino acids and screening the subsequent variants, substrate range and enantioselectivity can be engineered by sampling far fewer sequences. With these methods, it’s important to stress the first law of directed evolution: you get what you screen for.
Despite efforts made toward the design of optimal libraries, the first law of directed evolution also dictates it unlikely that all of the optimal sequences can be derived from a library if the synthesis of the library is flawed, as with traditional, but commonly used PCR based methods. This is a serious problem for protein engineers, as optimal variants may be completely missed, engineering experiments may completely fail to produce useful variants more often, and extensive screening requirements incur high costs per useful variant discovered. To avoid these issues, Professor Reetz’s paper stresses the importance of library synthesis methods that produce bias-free libraries if optimal engineering experiments are to be performed.
To this point, Professor Reetz’s paper compares the two key solid support library synthesis methods that have currently been applied to enzyme engineering in the contemporary literature: Sloning, and Twist Bioscience’s synthetic DNA manufacturing process featuring a high-throughput silicon platform.
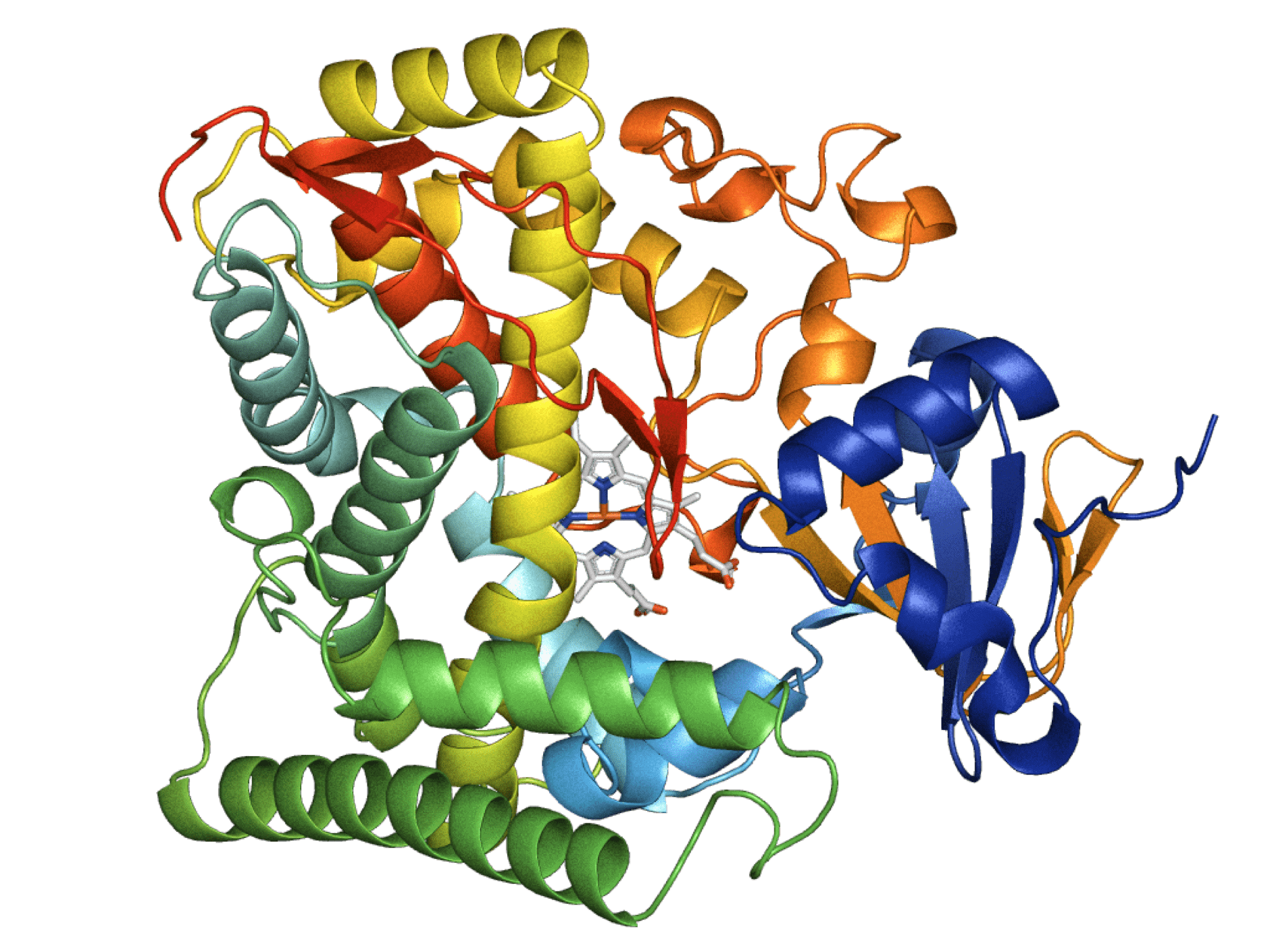
An example crystal structure of an enzyme from the P450BM3 monoxygenases. These are some of the most intensely studied biocatalysts in contemporary literature, and are a common target for bioengineering. This enzyme family was a target for engineering by Reetz’s lab, who used solid-phase sloning libraries to modify its enantioselectivity for testosterone. PDB: 1FAH. Image created using PyMol open source software. Image is available for use under CC BY-SA 3.0.
In previous work, Professor Reetz compared sloning libraries to PCR-based saturation mutagenesis libraries for the engineering of an enzyme that converts testosterone into a mixture of 2β-hydroxytestosterone and 15β-hydroxytestosterone. The study aimed to generate enantioselective enzyme variants that only produced one of the two testosterone products. Although the sloning library was found to contain considerably less bias than the PCR-based methodology, the paper also found a number of issues: “[Quality control] revealed problems such as multiple DNA-fragment insertions, wrong degeneracies, among other deficits.” Moreover, “the Sloning study revealed several quality-lowering characteristics which makes it statistically likely that amino acid bias was not fully eliminated.”
However, it must be noted that thorough statistical analyses of bias were not performed in these experiments and the extent of the bias in sloning libraries cannot be confirmed. In a more recent study, Reetz used massive sequencing to analyse the biases contained within a Twist Bioscience variant library, and an equivocal PCR-generated library for the engineering of the enzyme limonene epoxide.
Twist Bioscience uses the massive synthesis scale afforded by its silicon platform to directly synthesise every variant that has been designed by the researcher. To ensure exceptional quality of the library, Twist Bioscience then uses intensive quality control, including next generation sequencing to verify that the user’s design is represented by the library.
In Professor Reetz’s prior work, 97.3% of their designed diversity was obtained, with a near-perfect pattern of amino acid distribution after three rounds of screening, from which twice the number of useful variants were discovered than from a library produced by traditional PCR based methods.
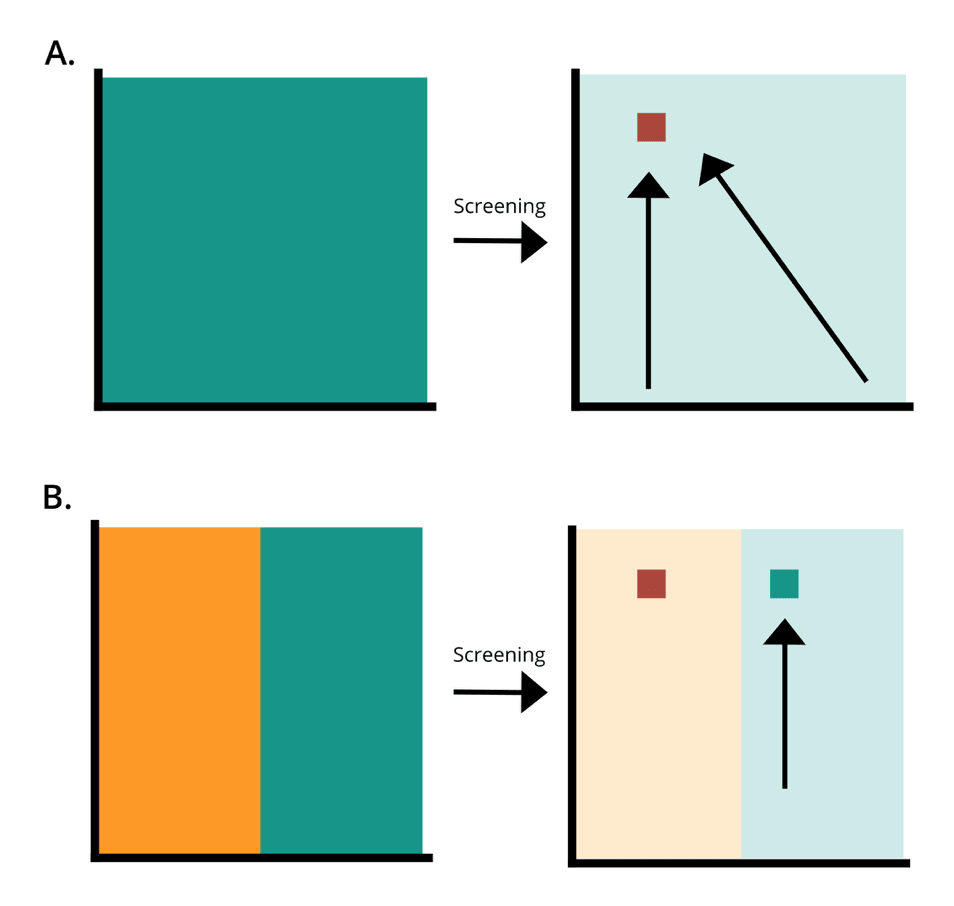
How better protein libraries allow for optimal protein design. A is a Twist Bioscience library. B is an equivalent PCR generated library. Green is the amount of the designed library present in the synthesised library. Orange is designed variation that is inaccessible through bias. The red square is the most optimal sequence for an application. By the first law of directed evolution “you get what you screen for,” if large proportions of the sequence space are inaccessible, protein engineering of libraries containing bias are highly likely to miss optimal sequences, causing experiments to fail.
Professor Reetz’s most recent review stresses how access to high-quality solid-phase libraries from Twist Bioscience enables the design of better enzyme engineering experiments. Twist Bioscience directly synthesises each individual variant in the library, which the paper notes provides the user freedom of design: “Remote randomization sites can be chosen, e.g., potentially for enhancing stability or for the mutational induction of allosteric effects. Here as before, the present approach can be expected to be superior to Sloning.” This means the user doesn’t have to constrain their designs to small blocks or single sites of saturation, and can be more holistic in their engineering approach.
By removing fabrication-induced biases, poor-quality variant libraries are no longer a factor in protein engineering experiments. Moreover, by freeing up library design, researchers gain experimental flexibility, allowing for multiple traits to be simultaneously screened, improving the workload required in engineering experiments.
Professor Reetz’s paper goes on to explain how the user is now able to focus on good library design, rather than the method of library synthesis, as the synthesis process is already taken care of: “The experimenter can then concentrate on optimizing the actual design of SM libraries [...] guided by structural data, bioinformatics, and molecular dynamics simulations. Subsequent to the proclamation of the first law of directed evolution—‘You get what you screen for’—the second law of directed evolution appears to be on the horizon: ‘You get what you designed’.”
Biocatalyst engineering using large-scale libraries is still a very young field. Professor Reetz’s lab is leading the way in the space by benchmarking the state-of-the-art tools that are available to protein engineers. PCR-based libraries will soon become a technology of the past, as solid-phase direct DNA synthesis of variant libraries is becoming increasingly available, provided by industry-leading companies like Twist Bioscience.
Featured image: Open source images of well-studied proteins from PDB

Twist Bioscience HQ
681 Gateway Blvd
South San Francisco, CA 94080