



 Oligo Pools
Oligo Pools Variant Libraries
Variant Libraries Synthetic Controls
Synthetic Controls







Get the latest by subscribing to our blog
In our previous blog pieces “Designer Enzymes with Beneficial Properties Generated Using Twist Bioscience Genes” and “Solid-Phase DNA Synthesis - The Future of Protein Engineering,” we discussed how protein engineering is an essential pursuit in today’s biological and biotechnological research. Engineering proteins provides the opportunity to expand our understanding of the molecular basis of life which enables the generation of better biocatalysts for industrial application and the development of new biological pharmaceuticals. Therefore, access to optimal technologies for protein engineering is a key driver of success in the field.
One such technology is site-saturation variant (SSV) libraries. SSV libraries are used for the discovery of single-point mutations that lead to altered protein function. The libraries consist of a complete set of protein variants, with each variant containing one of 19 possible amino acid variations at a single position of interest. In one library, it is possible to design any number of sites that can be saturated with variants.
Traditionally, SSV libraries are produced using polymerase chain reaction (PCR)-based methods such as error-prone PCR (epPCR), a technique that uses the imperfect nature of the DNA polymerase enzyme to introduce mutations throughout the protein-encoding DNA sequence. However, PCR-based methods provide researchers with little control over the production of their library, leading to the synthesis of an incomplete product that is hampered by sequence bias. When using these libraries, large screening efforts are required to ensure that the maximum possible variation is being analysed for mutations that harbor useful changes. Even then, researchers cannot be sure they are screening the complete set of variants and are unable to know if they missed any useful variants.
On the other hand, by virtue of high-throughput DNA synthesis developed by Twist Bioscience, the next generation of SSV libraries are now available. Every designed sequence variant is synthesised individually on Twist Bioscience’s patented silicon DNA synthesis platform, ensuring the presence of the maximum possible variation in the library without over or underrepresentation of single variants (bias). Before the library makes its way to the customer, it is also quality assured by next-generation sequencing.

The typical scheme employed when designing site saturation variant libraries
A recent paper published in ACS synthetic biology by David Ӧling and colleagues at AstraZeneca’s Innovative Medicines and Early Development Biotech Unit, collaborators at Imperial College London, and Twist Bioscience benchmarks Twist Bioscience’s SSV libraries as a tool for protein engineering. The paper focuses on the engineering of a G-protein coupled receptor from yeast that enables the fungus to sense glucose in its environment.
G-protein-coupled receptors (GPCRs) are proteins that sit in the plasma membrane of eukaryotic cells. Their role is critical for the cell’s ability to interact with its environment. In our bodies, our senses of sight, smell and taste are controlled by the activity of GPCRs. Additionally, they play key roles in the immune system and in the regulation of hormones, understandably, these proteins are of great pharmacological importance. For example, the disease retinitis pigmentosa which causes progressive blindness, is caused by mutations in the GPCR protein Rhodopsin. By gaining a better understanding of these diseases through research into their underlying molecular mechanisms, there is the potential to develop new pharmaceutical treatments. GPCRs are, as a result, a prime target for SSV library-based engineering.
The Ӧling paper is a proof-of-principle piece of research, that focused on a yeast GPCR that can be easily assayed for changes in its function. A total of 161 positions in the protein were predicted by the authors to harbour potential interactions with glucose (see image below). Every one of these positions were targeted for site saturation mutagenesis. The 3,059 variant-strong library was then synthesized by Twist Bioscience. To provide a comparison of library efficacy, an equivalent library was synthesised using epPCR. Both libraries were then subject to high-throughput DNA sequencing to analyze their quality based on three metrics: coverage, representation and stop codons.
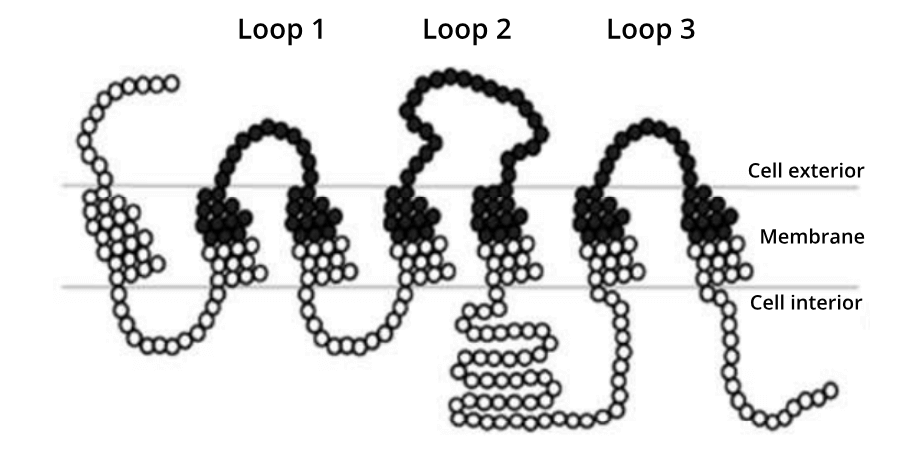
A simplified representation of the amino acids in the yeast glucose receptor. Black circles represent the 161 residues that Öling et al., 2018 targeted for site saturation mutagenesis.
Regarding the libraries’ coverage of designed mutants, the article reports: “Sequencing of the [Twist Bioscience] library confirmed the presence of 3055 of 3059 designed variants (99.9% representation) [...]. In contrast, the epPCR library contained 35% of the theoretical maximum of amino acid variants.” Having superior coverage in the Twist library provides researchers with the security that they are screening the maximum variation, ensuring increased opportunity for the discovery of mutations that produce new beneficial phenotypes.
Library representation relates to the proportion of each amino acid at a position in the library. In a perfect world, mutants would be evenly represented, ensuring that the screening process is not biased towards or away from certain variants. Even representation guarantees that minimal screening effort is required to capture the entire variation, saving time and cost. The article describes the ratios of amino acids present in the Twist library as “highly homogeneous,” whereas the variation in the epPCR library is described as “heterogeneous.” This variation is clearly evident in the heat map below, which shows that the Twist library possesses an almost uniform representation of variants across the board. On the other hand, the epPCR library has a number of residues that are considerably overrepresented (dark blue squares) compared to other residues at a given position which are in turn underrepresented.
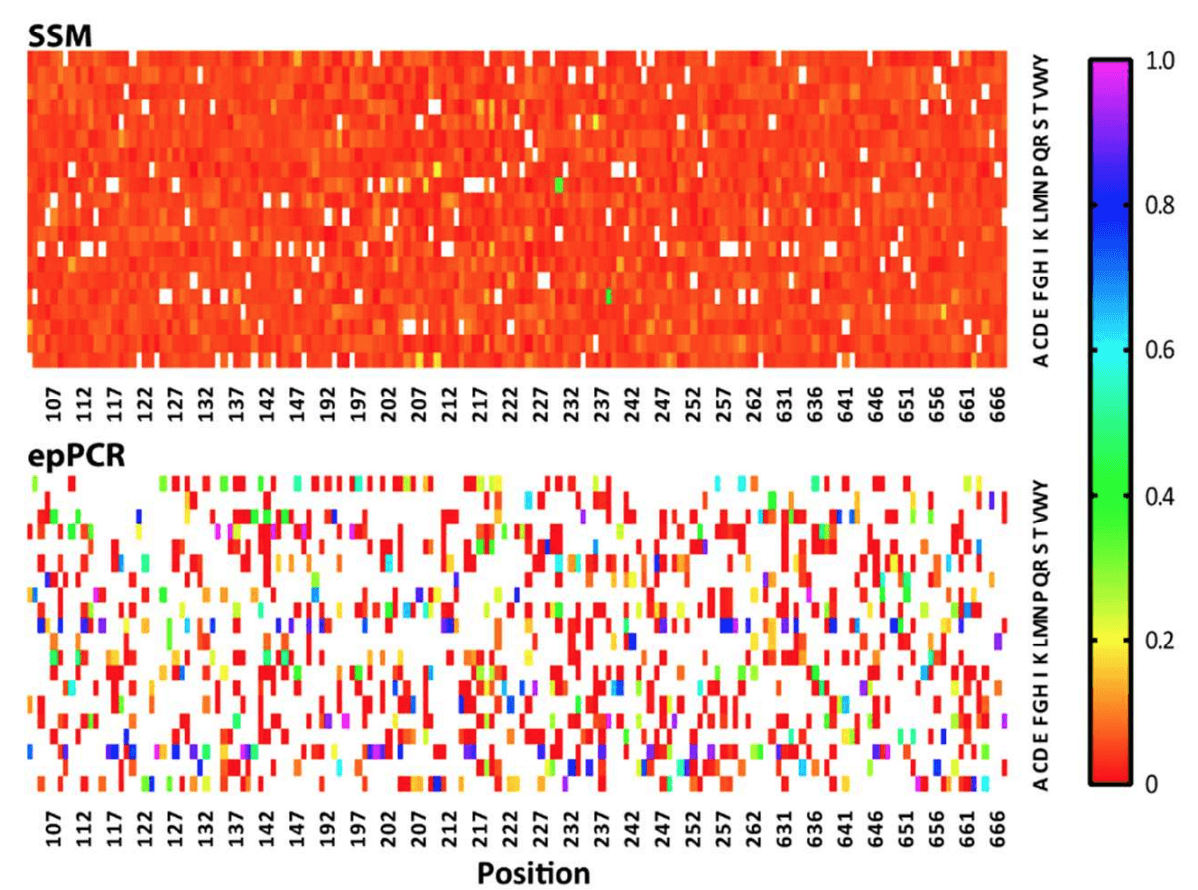
A heat map describing the quality of the two libraries tested by Ӧling et al., 2018. In the heat map, the X-axis represents the residue in the GPCR protein targeted for mutation, and the Y-axis represents the 20 possible mutations at each residue. White squares represent missing residues (each position should have 1 white square - the wild type). The scale represents the proportion of the mutations represented by given residue a given site.
Additionally, stop codons lead to useless, truncated proteins that are a waste of resources to screen. No stop codons are reported in the Twist library, however there is no comment made about the stop codons in the epPCR library.
The researchers then screened the libraries for mutants with increased activity using a simple fluorescence-based assay. Only a single beneficial mutant was discovered in the epPCR library. However, this same mutant alongside five more were discovered in the Twist library. Of the six variants found in the Twist library, one residue in the third extracellular loop gave the strongest observed increase in GPCR activity. Such an observation suggests that the residue may be an essential, previously undiscovered amino acid for controlling glucose response. To corroborate this finding, the researchers were able to look back through the Twist library and focus on just the variants at this single residue. As the researchers can be confident in the saturation of the Twist library, retrieving mutants of interest is simply an archiving task, which would not be possible with epPCR due to its poor coverage. When they looked at all variants at the site of interest, nine mutations were found to result in complete loss of GPCR function, whereas five led to an increase in function, confirming their hypothesis.
This study provides a clear proof-of-principal that Twist Bioscience’s SSV libraries are a highly optimized tool for the mutagenesis of multiple residues across a protein. Due to their superior synthesis process and NGS assured quality, cost-efficient libraries are now available to customers. Their complete, bias-free coverage also provides an ease of screening, further mitigating downstream expenses. In contrast, epPCR libraries are significantly limited in their representation and coverage, and lead to a number of issues during the screening process.
Given this data, variant libraries that are directly synthesized on Twist Bioscience’s silicon platform are set to enhance the future of protein engineering, allowing researchers to unlock the full variation in a protein’s sequence space. Using these libraries for the generation of better biocatalysts for bioindustry and for the development of new biological pharmaceuticals can only help to advance our understanding of the molecular mechanisms underlying genetic and acquired disease in the near future.
Featured image: Life science researcher grafting bacteria (Adobe Stock)

Twist Bioscience HQ
681 Gateway Blvd
South San Francisco, CA 94080