



 Oligo Pools
Oligo Pools
 Variant Libraries
Variant Libraries








{kind=link}
Get the latest by subscribing to our blog
Proteins are the molecular machines of life. Beginning as chains of amino acids translated from a corresponding RNA sequence, they acquire their functionality by folding into complex 3D structures. Nature makes the process look easy; even with millions of possible 3D configurations, most small proteins fold into a stable functional structure instantaneously through various atomic forces. Understanding how proteins fold is key to uncovering many sought after answers in biology. For example, how do mutations in the genome lead to disease, or how can an enzyme be engineered to effectively degrade plastic?
The ultimate question, a now 50-year-old postulate called the “protein folding problem,” asks whether a protein’s structure can be predicted using its amino acid sequence alone. Until now, the answer has been a firm NO. Given an amino acid chain, there are just too many possible structures to know which is correct.
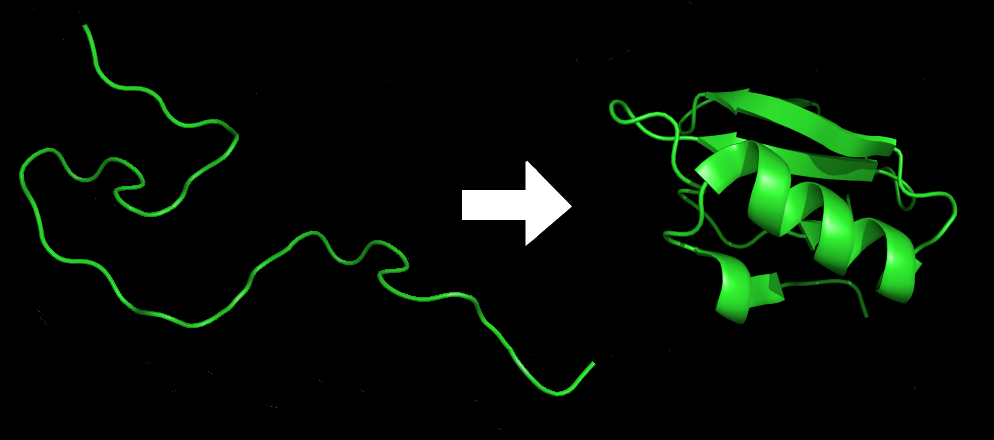
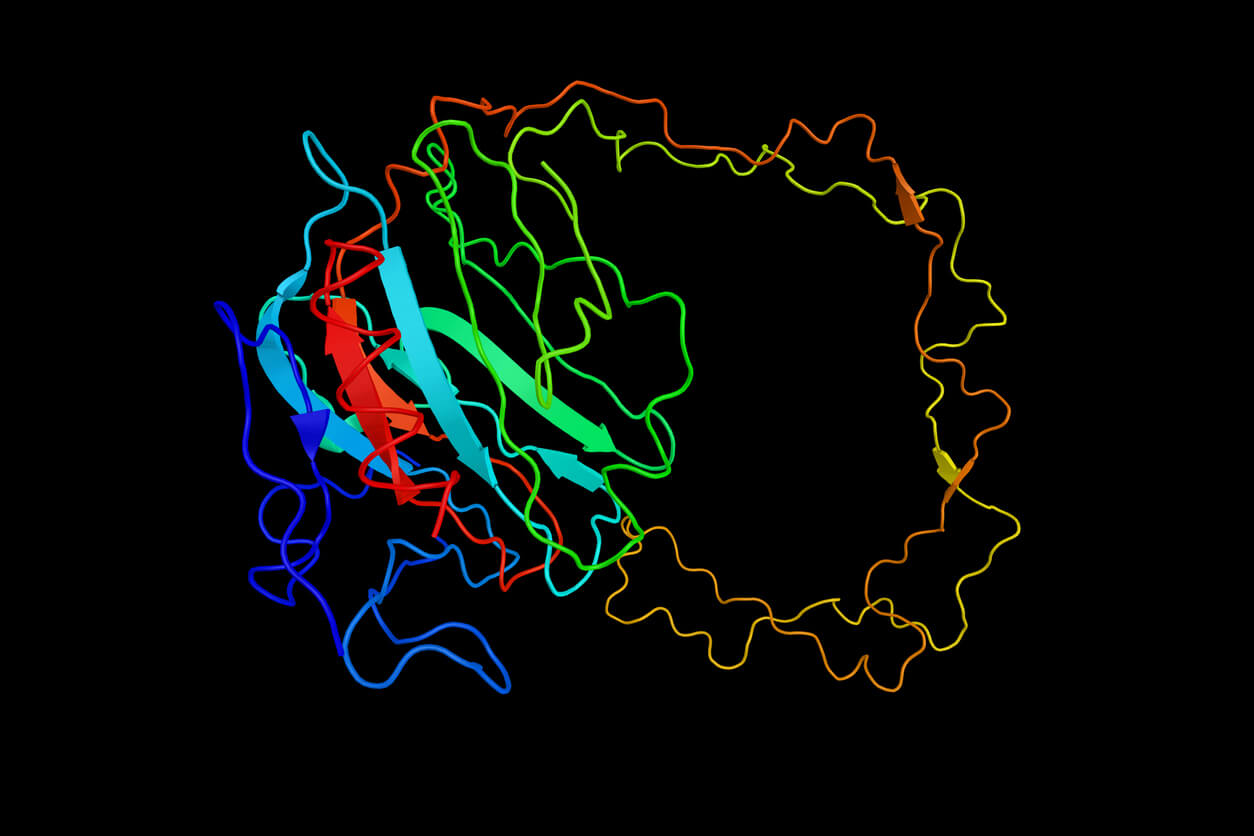
An amino acid chain for Chymotrypsin inhibitor 2 (1LW6) in its unfolded and folded form. Source: Wikicommons.
Typically, protein structures are determined by observing the diffraction pattern of X-rays passed through their crystallized forms. Blasting protein crystals with X-rays is easy; it’s making those crystals that is hard. Many call it a “black art”, as identifying the correct conditions to cause the nucleation of a protein crystal is again challenging, and typically achieved by brute force or pure luck. In the literature, examples even exist of stray eyelashes contaminating an experiment, but being the perfect seed for crystal nucleation. Alternative methods like cryo-electron microscopy and nuclear magnetic resonance spectroscopy have also been developed to solve protein structures. However, all of these lab-based techniques are both expensive and time-consuming.
That’s why scientists have been trying to devise computational methods for solving protein structures based solely on their amino acid sequence. And despite significant efforts, the field has been punctuated by small incremental advances in these methods for nearly 50 years – until now.
CASP competition Announces a Winner
Every two years, a conference is held to assess the latest developments in computational protein structure solving. Called the Critical Assessment of Techniques for Protein Structure Prediction (CASP), the biannual conference challenges teams to computationally solve protein structures that have only recently been determined with lab-based methods.
At this year’s iteration, CASP14, Google’s DeepMind blew the competition away with its artificial intelligence platform AlphaFold 2. Not only did it handily outperform its competitors, it did as well as lab-based techniques for over two-thirds of the protein structures it was given to solve.
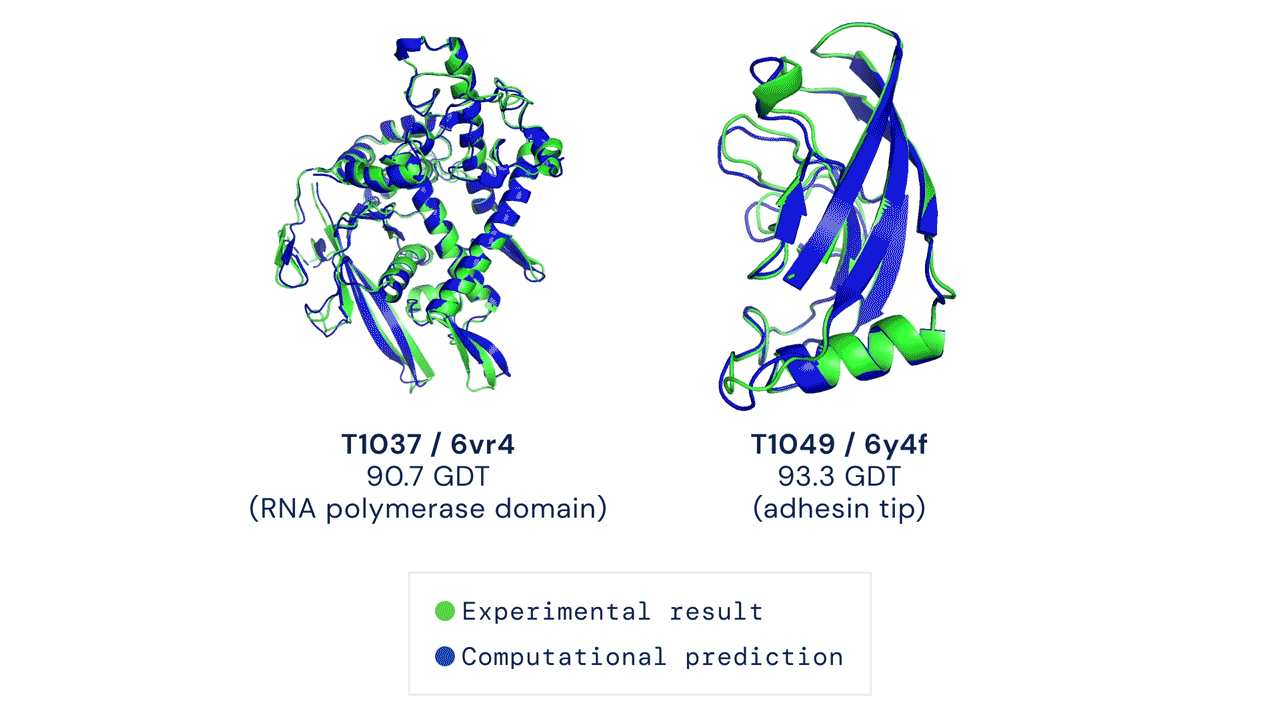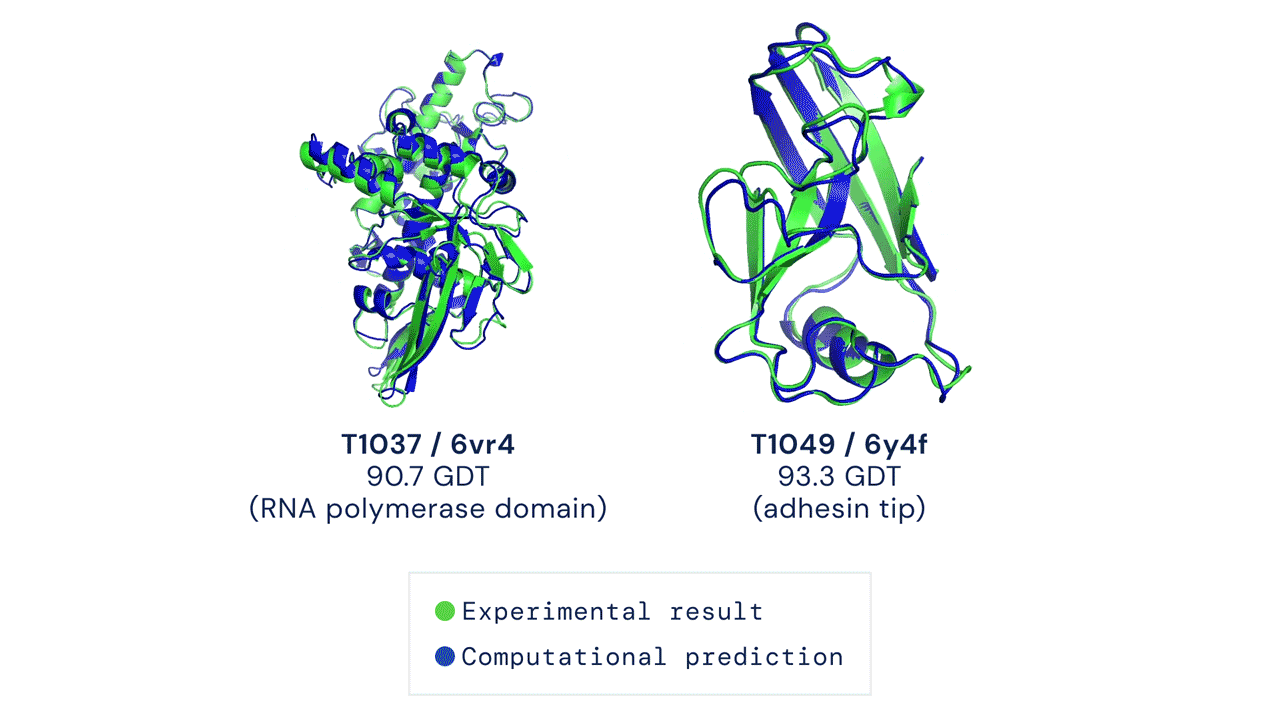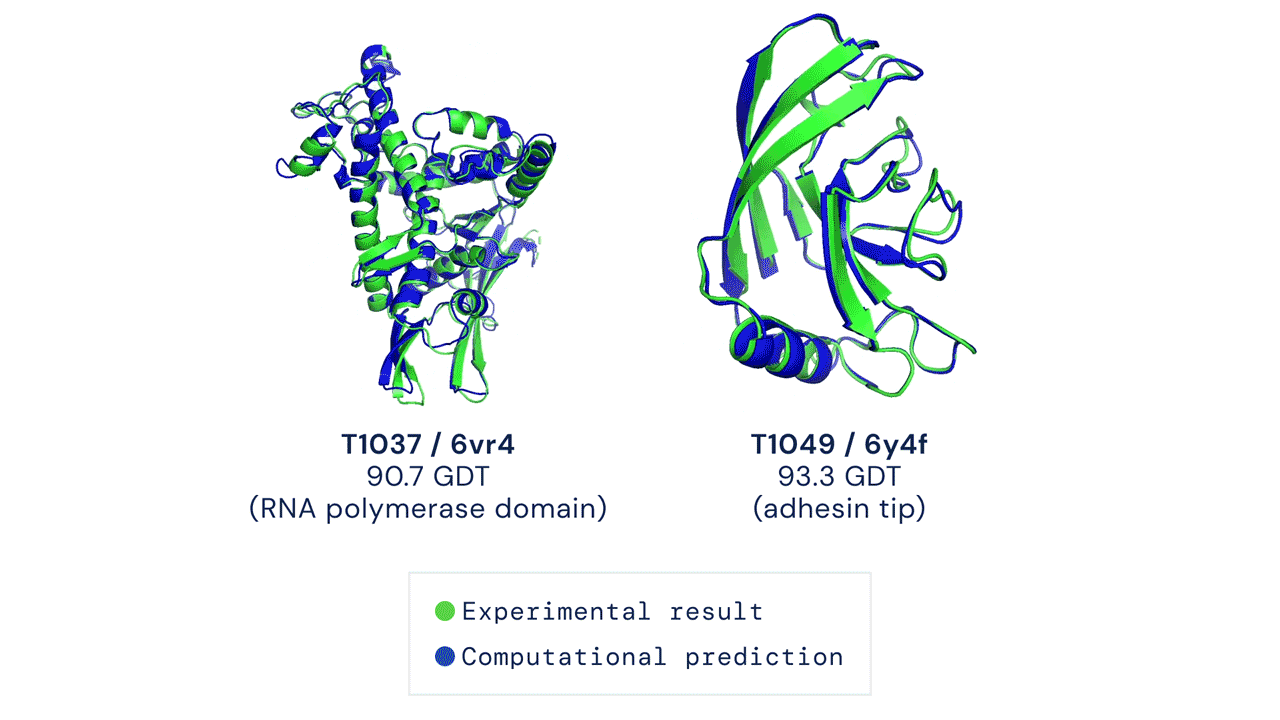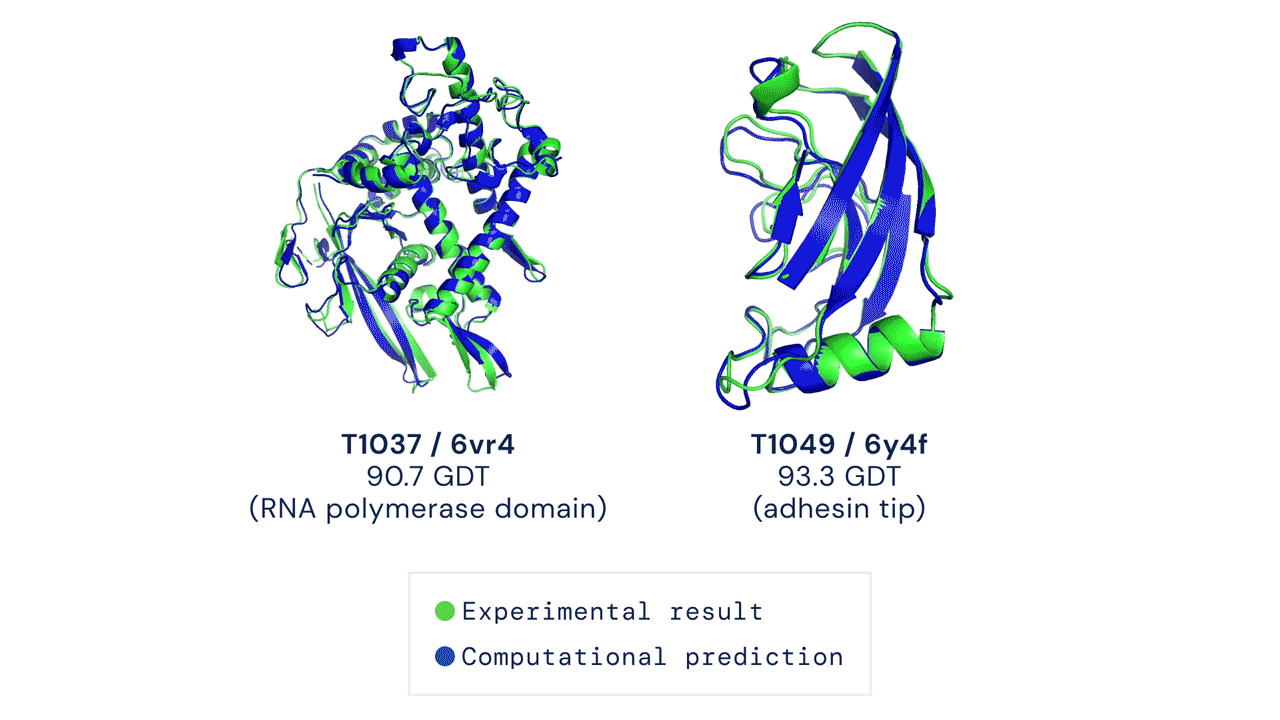
Two protein structures predicted by AlphaFold overlaid onto their experimentally deduced structures. Source
CASP measures the accuracy of protein folding algorithms with a metric called the Global Distance Test (GDT). Ranging from 0 to 100, the GDT essentially evaluates how close a computational prediction is to the structure determined in the lab. AlphaFold 2 scored a median of 92.4 GDT in CASP14. A score this high makes it difficult to pinpoint whether the computational or empirical structure is more “correct”.
Under the Hood of AlphaFold 2
The AlphaFold algorithm uses deep learning to solve protein structures. Deep learning is a form of machine learning that performs a task by automatically extracting the features of a system (e.g. 3D structure) from raw input data (e.g. many protein structures and amino acid chains). Although the inner workings of AlphaFold 2 won’t be published until early 2021, what’s clear is that it uses a deep-learning technique called an attention network.
A similar approach is used to train machines to understand language. Think of this blog article as a long amino acid chain. Just like the latter forms complex protein structures through a series of local and distant interactions between amino acids the former conveys meaning through local and distant relationships between words, sentences, and paragraphs. Attention-based deep learning algorithms synthesize these local and distant relationships to learn tasks such as understanding the meaning of language or determining the 3D structure of a protein.
In less abstract terms, AlphaFold 2 combined information derived from evolutionarily-related sequences, multiple sequence alignments, and interactions between amino acid residue pairs to learn how these variables are related across the 170,000 known protein structures used to train the algorithm.
Can AlphaFold 2 Solve Real-World Problems?
Its ability to solve structures in weeks rather than months (or years) points to a role for the AI platform in therapeutic small molecule discovery and the engineering of new proteins for a variety of real-world applications, from optimizing industrial processes to degrading plastic.
Computational methods are frequently used to determine how drugs interact with their protein targets. Unfortunately, the structure of thousands of druggable proteins remains unknown, making it difficult to develop effective drugs against them. Predictive algorithms like AlphaFold 2 are promising solutions to this backlog if shown to be consistently accurate. In a proof-of-principle demonstration, AlphaFold 2 accurately predicted the structure of the SARS-CoV-2 Spike protein, the main therapeutic target against COVID-19, earlier this year. It also predicted several other SARS-CoV-2 proteins before they were confirmed by lab-based methods.
Another game-changing application where AlphaFold 2 may make its mark is in protein engineering. Often called the inverse protein-folding problem, protein design aims to identify amino acid sequences that will form stabilized, functional, useful protein structures. It encapsulates everything from engineering receptors with different ligand specificities, enzymes with altered activity, and improved biocatalysts to designing entirely new proteins. The former three examples are simpler in that they modify existing proteins by figuring out which amino acids to change. Completely de novo protein design (building entirely new proteins from scratch) promises a near unlimited palette of chemical reactions, biological interactions, and receptor cascades at our disposal. In fact, some AI algorithms have already demonstrated some success in this arena.
Twist Gene Fragments and Variant Libraries Facilitate such Protein Engineering challenges
Protein engineering applications currently benefit from Twist’s suite of products for protein expression and mutagenesis. These efforts are epitomized by the Baker lab’s efforts to design new protein therapeutics against botulism and influenza. By combining computational tools and Twist Oligo Pools, Baker’s team was able to greatly accelerate the design-build-test cycle of de novo protein engineering. Twist also offers Gene Fragments that simplify the cloning process for small-scale protein expression experiments and Variant Libraries that enable high-throughput screening of mutant proteins with full variant representation.
Whether or not AlphaFold 2 could supplement existing efforts in protein design, or solve the inverse protein-folding problem altogether, remains an open question. AlphaFold 2 did not match the performance of lab-based methods for roughly a third of the structures assigned by CASP14, indicating room for improvement. Nevertheless, AlphaFold 2’s overall performance far surpassed its competitors and that of its 2018 predecessor. If that’s the performance improvement we should expect in just two years, then we can only imagine what the next iteration will be capable of.

Twist Bioscience HQ
681 Gateway Blvd
South San Francisco, CA 94080