Using Synthetic Biology to Engineer Membrane Proteins
Synthetic DNA libraries have been used for many years to generate multiple versions of proteins to test.
Synthetic DNA is changing the way research is performed. Utilizing state-of-the-art synthetic biology methods, large numbers of constructs can be rapidly created, allowing scientists to focus on performing their downstream experiments and accelerating their discoveries.
Engineering Protein Function with Synthetic Biology
Synthetic DNA libraries have been used for many years to generate multiple versions of proteins to test. Researchers can order variant libraries containing many different sequence combinations to explore the possibility of improving the binding properties of antibodies or the catalytic activities of enzymes.
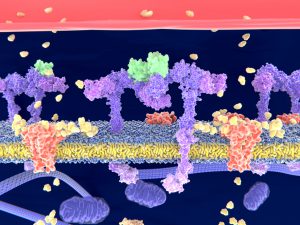
Membrane proteins are involved in many aspects of cellular biology, for example, they regulate how cells interact with their environment and thus are important drug targets. Until recently, they have not been investigated to the same extent as other soluble (non-membrane) proteins because their inherent properties present some unique difficulties.
Membrane proteins have extracellular, membrane spanning, and intracellular domains, allowing cells to sense stimuli and interact with the environment.
Integral membrane proteins have a portion of their amino acid sequence spanning a cellular membrane, which is challenging to correctly reproduce in many experimental settings, often making it difficult to express and localize these proteins. Because their complex structures consist of extracellular, membrane spanning, and intracellular domains, changing the sequence of membrane proteins to investigate function often results in non-functional proteins. Consequently, it is more difficult to engineer membrane proteins by standard mutagenesis methods, relative to other classes of proteins.
When Standard SynBio Doesn’t Work
Dr. Frances Arnold’s lab at the California Institute of Technology has developed a creative solution to studying and engineering complex proteins that they call “structure-guided SCHEMA recombination”. SCHEMA refers to a computational algorithm that scores a protein based on its three dimensional structure and identifies specific “blocks” of the protein that are likely to work together as a unit within the larger structure. The corresponding blocks can then be swapped between functionally similar but sequence-diverse proteins. The resulting chimeric proteins are studied to investigate the contribution of each block to the overall protein function. This approach allows features of different proteins to be combined to engineer new proteins with functional improvements.
The structure-guided SCHEMA recombination method developed by Frances Arnold’s lab at Caltech identifies blocks within a protein that likely to function together, allowing the researchers to swap these blocks between proteins with diverse sequences, but similar functions, to create a libraries of chimeric proteins. Figure provided by the Arnold lab.
Structure-guided SCHEMA recombination has been used previously to successfully engineer several soluble proteins, including β-lactamase and cytochrome P450, but has only recently been applied to membrane proteins. In this new report by the Arnold lab in the Proceedings of the National Academy of Sciences, the authors applied their structure-guided SCHEMA recombination approach to create libraries of chimeric light-responsive membrane protein channels.
The light-gated protein channels, called channelrhodopsins (ChRs), have the ability to sense light. When activated by light they open an ion channel, allowing positive ions to flow into the cell. ChRs are present in microorganisms such as photosynthetic algae but can also be expressed in neurons to stimulate action potentials (firing of neurons) to allow genetically-encoded control and regulation of neuronal activity.
Three dimensional structure of a chimeric ChR protein. Figure provided by the Arnold lab.
Optimizing Light-Gated Channels
The authors selected three different and diverse parental ChRs for their study. These parental proteins share 45-55% amino acid sequence identity, which is representative of the sequence diversity within the ChR family of membrane proteins. The SCHEMA algorithm identified two recombination libraries, each with 10 different blocks within the parental ChRs. One contained contiguous sequence blocks while the other contained blocks that consisted of amino acids in close proximity in three-dimensional space, but not necessarily next to each other in the primary linear sequence. Together, the libraries comprise over 100,000 variants.
From these, the researchers chose a set of >200 different combinations of the blocks. This set included chimeras generated by swapping each single block of one parental protein with the corresponding block from the other two parents, as well as other specific combinations selected for testing. This experimental design allowed screening of many different naturally-occurring sequence variants in different combinations.
Twist Bioscience synthesized the set of >200 sequences as a collection of NGS-verified clonal genes in mammalian expression vectors using its proprietary silicon-based DNA writing technology. Because the sequences in the library were synthetic, and do not exist in nature, it would have been very difficult and time-consuming to generate them using traditional molecular cloning methods.]
Three Arnold lab researchers leading this work: Austin Rice (left), Claire Bedbrook (middle), and Kevin Yang (right).
According to Claire Bedbrook, a graduate student in the Arnold lab and the lead author on this report, “The sequences we designed were very difficult to work with, and the only way we could have completed this project was to have them synthesized and cloned by Twist Bioscience.”
The final set consisted of 215 designed sequences synthesized by Twist Bioscience, in addition to the three parental ChR sequences. Dr. Emily Leproust, CEO of Twist Biosicence, and Dr. Siyuan Chen, director of chemistry and molecular biology at Twist Bioscience, were co-authors on the paper.
Eighty-nine percent of the chimeric sequences chosen were expressed to higher levels than the lowest expressing parent. However, only 44% of these chimeric membrane proteins were localized to the correct membrane position with higher efficiency than the lowest efficiency parent sequence. Furthermore, there was no observed correlation between expression levels and correct localization, leading the authors to conclude that there are different sequence requirements for expression levels and correct intracellular localization.
Data generated by the Arnold lab showing the range of chimera protein expression (left) and correct localization (right), relative to the the parental proteins. Figure provided by the Arnold lab.
There was not a clear pattern specifying which sequence block contributed most to expression and localization, suggesting that different blocks can contribute context-dependent properties. The contiguous and non-contiguous libraries exhibited similar ranges of results, validating that a dual approach is likely advantageous when performing protein engineering experiments.
Finally, the chimeric membrane proteins that localized correctly were investigated for other functional properties, such as responses to light. More than 90% of the properly localized chimeras were activated by light, but they varied in the wavelength of light that resulted in maximal activation. Additionally, several of the chimeras exhibited a stronger response than any of the parental ChR membrane proteins, indicating that chimeric proteins could possess new or improved activities.
What’s Next?
Now that the researchers have investigated which sequences in the chimeric ChRs result in higher expression levels and improved localization, they are implementing machine learning algorithms to extend the analyses to help predict what other specific changes could be made to produce desired results.
“As a next step,” Claire Bedbrook explains, “we have used our initial data set to train computational models that allow us to accurately predict which amino acid sequences control expression, localization, and other functional activities. We have already validated this approach by generating new sequence variants whose experimental behavior matches the predictions of our models, and are looking forward to expanding our research by continuing to work with Twist Bioscience to have many more sequence variants synthesized.”
This has exciting implications for the future of protein engineering. Being able to more precisely predict the functional consequence of changing protein sequences would allow synthetic biologists to reprogram biological machines to efficiently perform many different activities, such as more sustainable chemical synthesis, generation of alternative fuels, and production of novel therapeutic compounds.
Frances Arnold is a member of the Twist Bioscience Scientific Advisory Board.




 Oligo Pools
Oligo Pools
 Variant Libraries
Variant Libraries