



 Oligo Pools
Oligo Pools
 Variant Libraries
Variant Libraries








Get the latest by subscribing to our blog
Protein engineering can seem like an insurmountable task when you look at the numbers. For a protein with 100 amino acids in length, each position could be altered in 20 different ways, meaning the total “sequence space” for this protein contains 1021 possible variations. If you counted every grain of sand on earth, you would still have less than 1/100th of this huge number. Also, due to the dynamic and unpredictable structure-function relationships in proteins, there is simply no way of knowing which one of these amino acid variations will give optimal activity for the engineering task at hand.
To solve this number problem, protein engineers adopt nature’s own engineering tools, looking to the principles of evolution to push sequences toward favorable properties.
Professor Manfred Reetz, Professor Emeritus at the Max Planck Institute für Kohlenforschung in Mülheim, Germany, and Marburg University is one of the forefathers of protein engineering, best known for his pioneering work on directed evolution. His lab just published a groundbreaking study that could be a game-changer for the protein engineering field. We had the privilege of discussing the work with Professor Reetz, and with his help, we were able to dive into the finer details of his study.
Titled “Beating Bias in Directed Evolution of Proteins: Combining High- Fidelity On-Chip Solid-Phase Gene Synthesis with Efficient Gene Assembly for Combinatorial Library Construction,” the paper characterizes two directed evolution-driving technologies in more detail than ever before. By using massive sequencing, his team was able to assess how the polymerase chain reaction (PCR) causes bias in the directed evolution workflow, damaging the experiment. They also showed that these biases could be solved by generating the libraries by direct synthesis instead.

Professor Manfred Reetz (right) with the lead author on the paper Dr. Aitao Li.
Directed evolution is a simple technology in principle. In nature, mutations in an organism’s genome cause alterations to protein amino acid sequences. Slowly, mutations that cause slight improvements in an organism’s ability to survive in its environment become refined in a population. Simply, evolution optimizes a protein to function optimally in their natural setting over millions of years.
Directed evolution allows researchers to take control of this steady optimizing force, and expedites it massively in the laboratory for the engineering of proteins. First, researchers create a library of protein mutants simulating genome mutation at a huge scale. Depending on the nature of the engineering experiment, there is the possibility to create up to 1010 mutants, each a unique variant on the natural protein sequence. The variant library is then subject to intensive screening, and the mutants with the most optimal function for their required roles are extracted. It’s Darwin’s “survival of the fittest” concept condensed into a test tube.
“Screening, not creating these variants is the bottleneck inefficient engineering,” stated Professor Reetz when we asked him about this process. “If we can make smaller numbers of variants but still get to the same result, then we have to screen less.” His lab specializes in developing protocols that make library generation highly efficient and take the difficulty out of the screening process. By using the protein structure to guide decisions on exactly which amino acids at which positions in the protein could produce beneficial phenotypes, small, highly optimized variant libraries can be generated.
Importantly, when producing any size of protein variant library, every possible variant should be represented within the pool of designed sequences. If sequences are missing at the DNA level, optimal mutants could be completely missed at the protein level. Additionally, having an unequal representation of variants within the library is damaging to the screening process. Certain sequences become common, and others rare, greatly decreasing the likelihood that some would be captured in the screening process. Again this means that potentially beneficial mutations are likely to be missed in such a biased library.
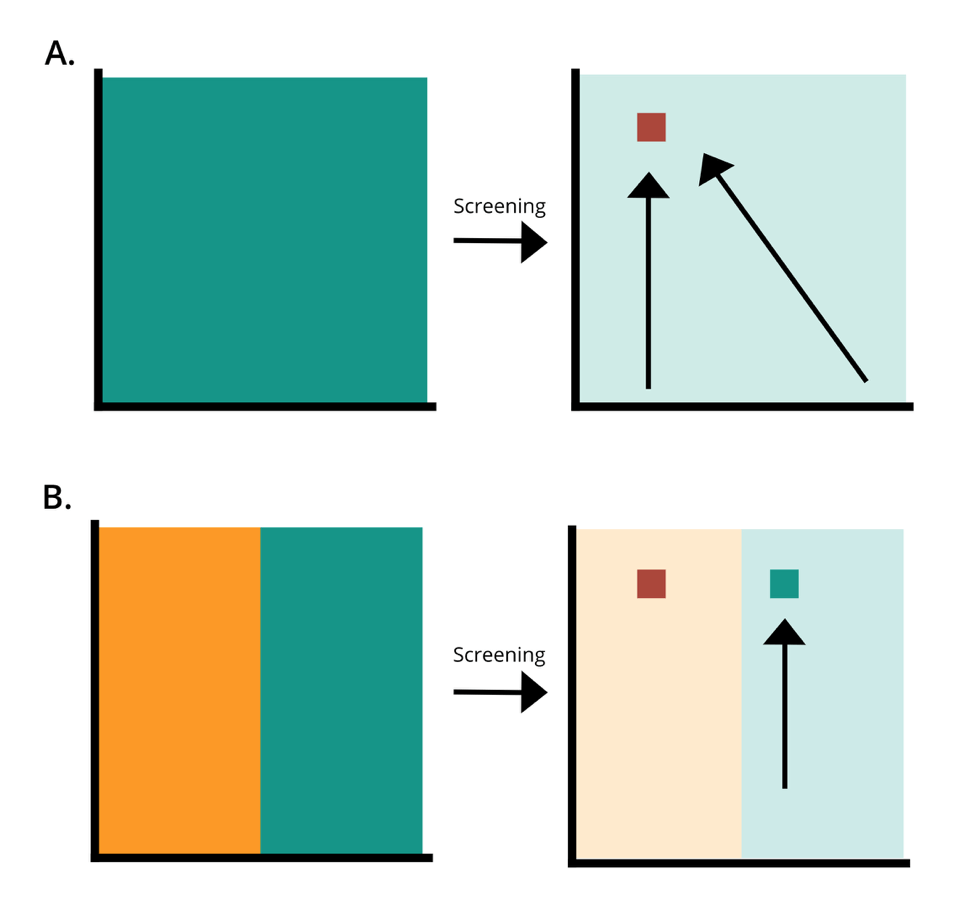
“It has been known for many years that PCR causes bias in the mutant library construction. Codon redundancy and poor quality of the primers you purchase are all problems,” commented Professor Reetz. “However, no one knew the extent of these problems.” In the literature, it may look like this bias isn’t much of an issue—there are many publications that harbor improved mutants developed from PCR-based libraries. Yet, the nature of scientific publication means it is often only the experiments that work that gets published.
Importantly, there is a distinction between a variant that is sufficient for the desired function and a variant that is optimal for a function. If a protein is used as a catalyst in industrial processes, sufficient vs. optimal could mean a difference of millions of dollars in revenue. Reetz’s lab set out to understand the impact this unmonitored bias may have on protein engineering while searching for a solution.
Their directed evolution target was a bacterial enzyme called limonene epoxide hydrolase (LEH)—an enzyme that converts the citrus-derived molecule limonene into a mix of chemical enantiomers in a 48:52 ratio. Enantiomers are two molecules of the same atomic composition, that are mirror images of one another. Your hands are enantiomers—they look the same but lay them palm down one over the other and they have clearly different symmetry.
Enantiomers are a big deal in the chemical and pharmaceutical industries, as their differences in symmetry cause different chemistries. A famous example is the drug thalidomide. One enantiomer cured morning sickness during pregnancy, but the other caused serious birth defects. In the chemical industry, separating enantiomers can come at a great cost, so enantioselective enzymes are highly valuable alternatives.
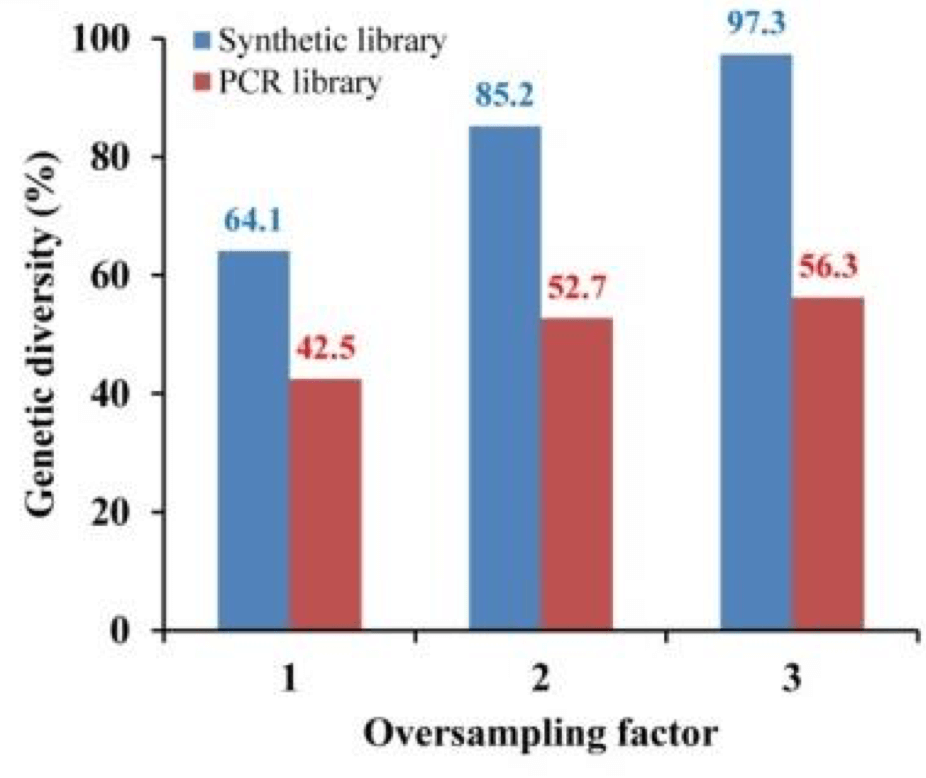
Reetz’s lab built two identical variant libraries, one by PCR and one constructed with brand new technology developed by Twist Bioscience that directly synthesizes every variant on a silicon surface. The two libraries contained four possible amino acid variants at four positions in the protein’s active site giving 256 possible variants. The researchers then oversampled the two mutant libraries 3x (768 mutants per library) and then massively sequenced the mutants they sampled to understand the libraries’ composition in fine detail.
“To my complete surprise, the PCR library only had about half [56%] of the total genetic diversity. It was impossible to get all the mutants. This was eye-opening for me,” Reetz said of his findings. On the other hand, the library generated by solid-phase synthesis performed dramatically better, containing over 97% of the designed sequences.
 ;
;
When considering amino acid representation within both libraries, a similar trend appeared. Where the synthesized library contained an almost even distribution of amino acid variants at each site, PCR-based libraries did not. Instead, the variants were considerably biased toward the original amino acid composition of the protein, and failed to evenly distribute amino acids throughout the library.
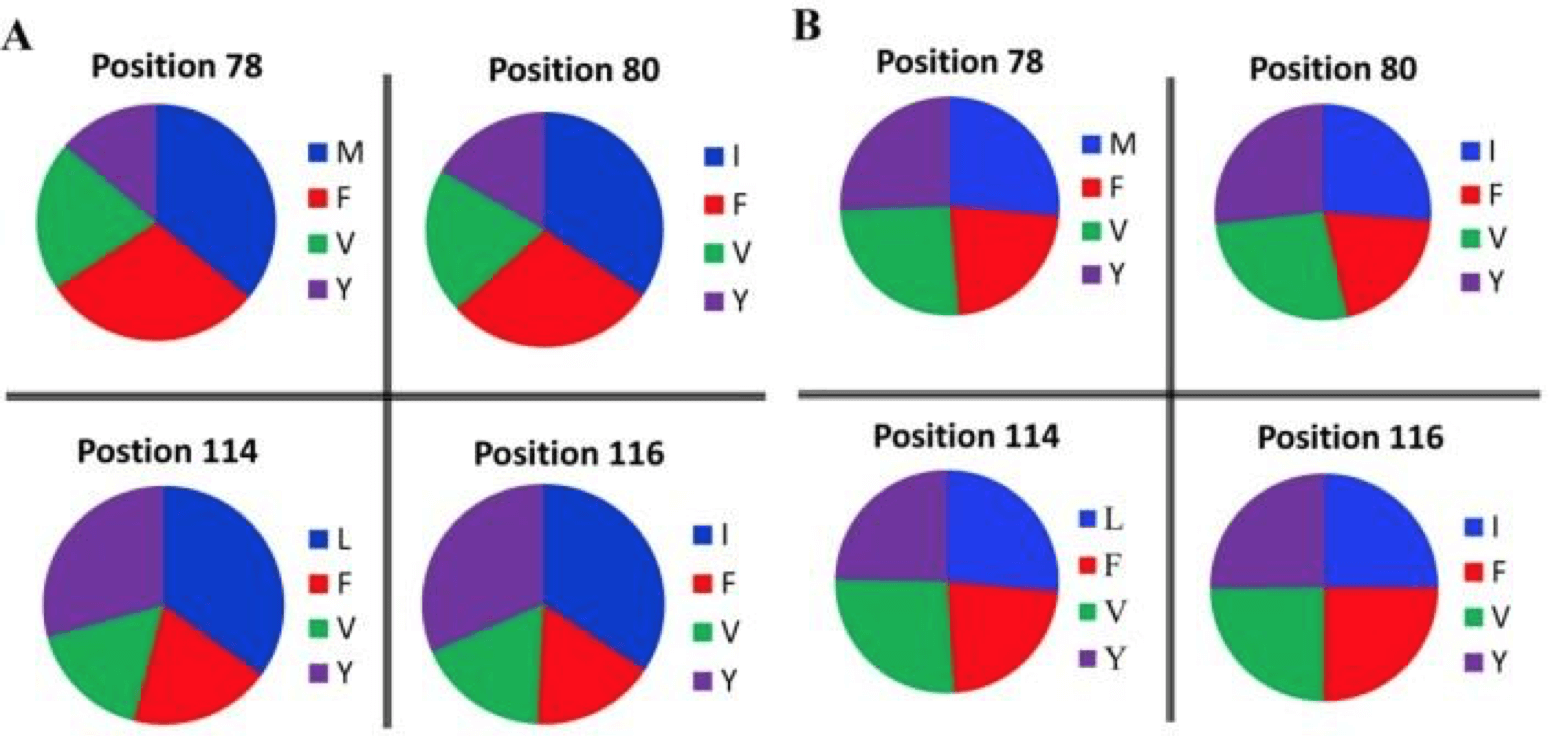
In fact, in every condition the researchers tested, the directly synthesized library came out on top. It was highly accurate to the researcher’s design, and when the mutant LEH’s properties were tested, 17 highly enantioselective variants were discovered. This was over twice as many than were identified in the PCR-based library, which only harbored 8. For the first time, Professor Reetz’s team could quantify exactly how much bias impacted variant libraries when they were constructed by PCR.
It must be added that engineering enantioselectivity is only the first step in engineering an industry-ready enzyme. “We did not measure activity nor any tradeoff in thermostability,” comments Professor Reetz. However, from the synthesized library, his team now has a much greater pool of enantioselective sequences available to screen for such properties.
When we asked Professor Reetz for his thoughts on any downsides of his experience with the synthesized library, he had this to say: “It should not be ignored that the synthetic on-chip library was not absolutely perfect, a very small amount of bias remained. While future research may possibly reduce bias even further by focusing on the synthetic steps or the final assembly, 100% elimination is not necessarily required in practical applications, simply due to statistical reasons.” He further added that caveat that his study is still a “proof-of-principle” and more work needs to be done in the future.
He also commented that in the future, protein engineers need to pay greater attention to the detrimental effects of using PCR-based libraries over more modern techniques. Alternatively, if they are using PCR-based libraries, they need to at least assess the damaging effects of bias to make better-informed decisions about how they can go about screening the library to make the most out of the library.
Twist Bioscience is a market leader in high throughput DNA synthesis, able to fabricate bias-free variant libraries by directly synthesizing the variants onto proprietary silicon chips. "In the future, hopefully, the [library] customer will not have to perform any internal quality controls, and they can just buy a library and trust that it is of exceptional quality. Twist Bioscience provides a major step forward in gaining that trust,” finished Professor Reetz. Directly synthesized DNA libraries will soon be the tool of choice in the protein engineer’s toolbox.
For the first time, a window into the vast benefits of synthetic on-chip libraries in protein engineering has opened. Higher quality libraries, mean easier screening, fewer man-hours wasted, and an increased number of discoveries in every library. Simply put by Professor Reetz: “You get what you design.”

Twist Bioscience HQ
681 Gateway Blvd
South San Francisco, CA 94080