Exome Sequencing 101: Part 3 - Next-Generation Sequencing (NGS)

Exome Sequencing 101: Part 3 - Next-Generation Sequencing (NGS)
We are upon the third installment of the Exome Sequencing 101 blog series. In the last two parts of this series, we walked through the preparation of a genomic sample into an adapter-tagged library, and followed up with the processes behind target enrichment that allow researchers to isolate just the 1% of the genome that encodes the exome. In this piece, we will explore how a refined genomic sample is read and converted into a digital data file that allows researchers to unlock the sequence of the genome.
In 2003, the first draft of the 3.3 billion base pair human genome was published by the Human Genome Project. This first read through was conducted mainly with a low-throughput method called Sanger sequencing, and the cost was close to $3B. Today, high throughput next-generation sequencing (NGS) technologies have improved, enabling researchers to sequence a whole human genome for around $1,000. Now, nearly all genome sequencing is run on a NGS system.
There are numerous NGS platforms that have been developed, each using unique methods to read DNA in huge volumes. The most widely used commercially available methods are:
Illumina (solexa) sequencing by synthesis: Enzymes grow a lawn of short single stranded DNA molecules into double stranded DNA. Fluorescent molecules fill the single strand in to make a double stranded molecule. Sequencing machines read the fluorescent signal from each new base as it’s being added to build up the sequence of the strand.

Ion Torrent semiconductor sequencing: As short single strands of DNA are extended by an enzyme into double stranded DNA, each new base added releases a proton, changing the acidity of its surrounding environment. By flooding the sequencing environment with a single type of nucleotide, only the DNA strands that are able to incorporate the nucleotide show a detectable acidity change. Repeating sequentially reveals the sequence of every DNA strand in the sequencer.
PacBio Single Molecule Real Time sequencing: DNA is fed through an enzyme called DNA polymerase that is fixed to the bottom of a well. The well is filled with fluorescently tagged nucleotides and each new base that is added is monitored with a concentrated light source.
Nanopore sequencing: As DNA is threaded through nanometer-wide pores in a membrane, each base causes a unique fluctuation in the ionic current across the pore which can be measured.
Around 90% of all DNA sequencing data is produced on Illumina systems. Illumina sequencers make short reads of up to 300 base pairs (bp), but do so in massive parallel. Our hypothetical patient sample from the previous two posts has already been fragmented, had Illumina-specific adapters added, and had just the exome isolated by enrichment.
Now the sample is ready to be sequenced using an Illumina sequencer, such as the HiSeq4000.
Loading
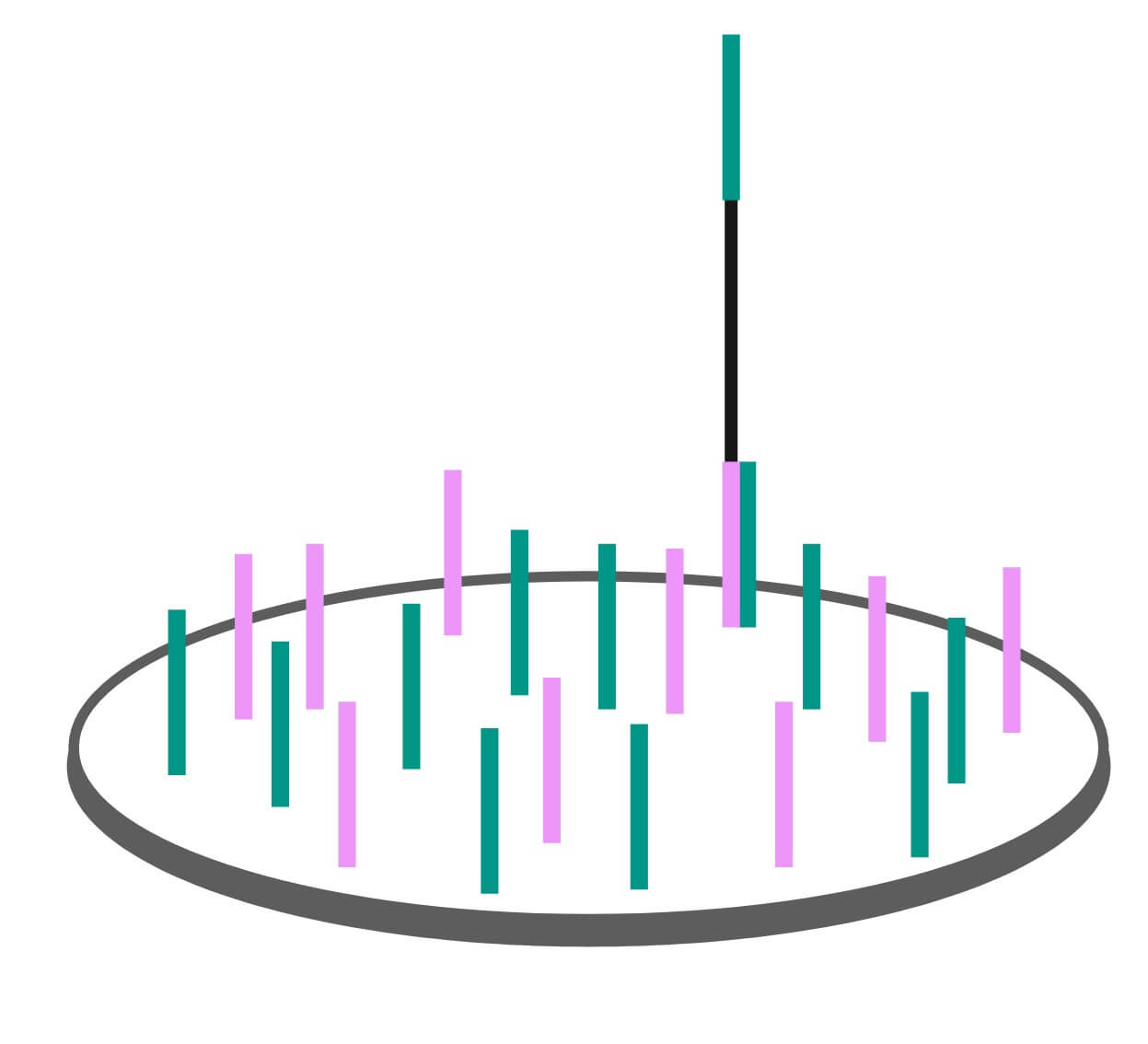
The first step requires the researcher to load the DNA sample onto a flow cell. The flow cell is a device patterned with billions of nanometer sized wells. Inside each well are small single stranded DNA oligomers that are affixed to the well surface.
In the Exome Sequencing 101: Library Preparation post we described how each genomic DNA fragment is ligated to an adapter. The DNA strands affixed into each well on the Illumina flow cell are a complimentary match to these adapters. They will capture the adapters attached to the single stranded genomic DNA fragments that make up the exome enriched genomic library as they are washed over the wells. The flow cell and sequencing preparation process are designed such that only a single genome fragment is captured per well. This is vital, as the sequencer reads the DNA sequence at the well level. If there is more than one fragment, the signal from both sequences gets mixed together and produces poor quality sequence data.
Clustering
Once the genomic fragments have been captured in the wells by their adapters, the Illumina sequencer begins a process called clustering. One DNA fragment in a well wouldn’t produce a strong enough signal to measure, so an enzymatic process called bridge amplification multiplies the single bound fragment. The resulting double stranded fragments are denatured, and the process repeats to produce a cluster of millions of identical bound single stranded fragments ready to be sequenced. We spoke to Mark Consugar, Field Application Scientist for Twist Bioscience, about this process and he gave the following analogy:
“Imagine a solitary tree in an empty field. If you were to visit that tree 20 years later, there would be a coppice of trees surrounding it as it dropped its seeds. Another 20 years and all these trees have also dropped seeds. Before long the field has turned into a forest. In principle, this is how bridge amplification multiplies a genomic fragment.”
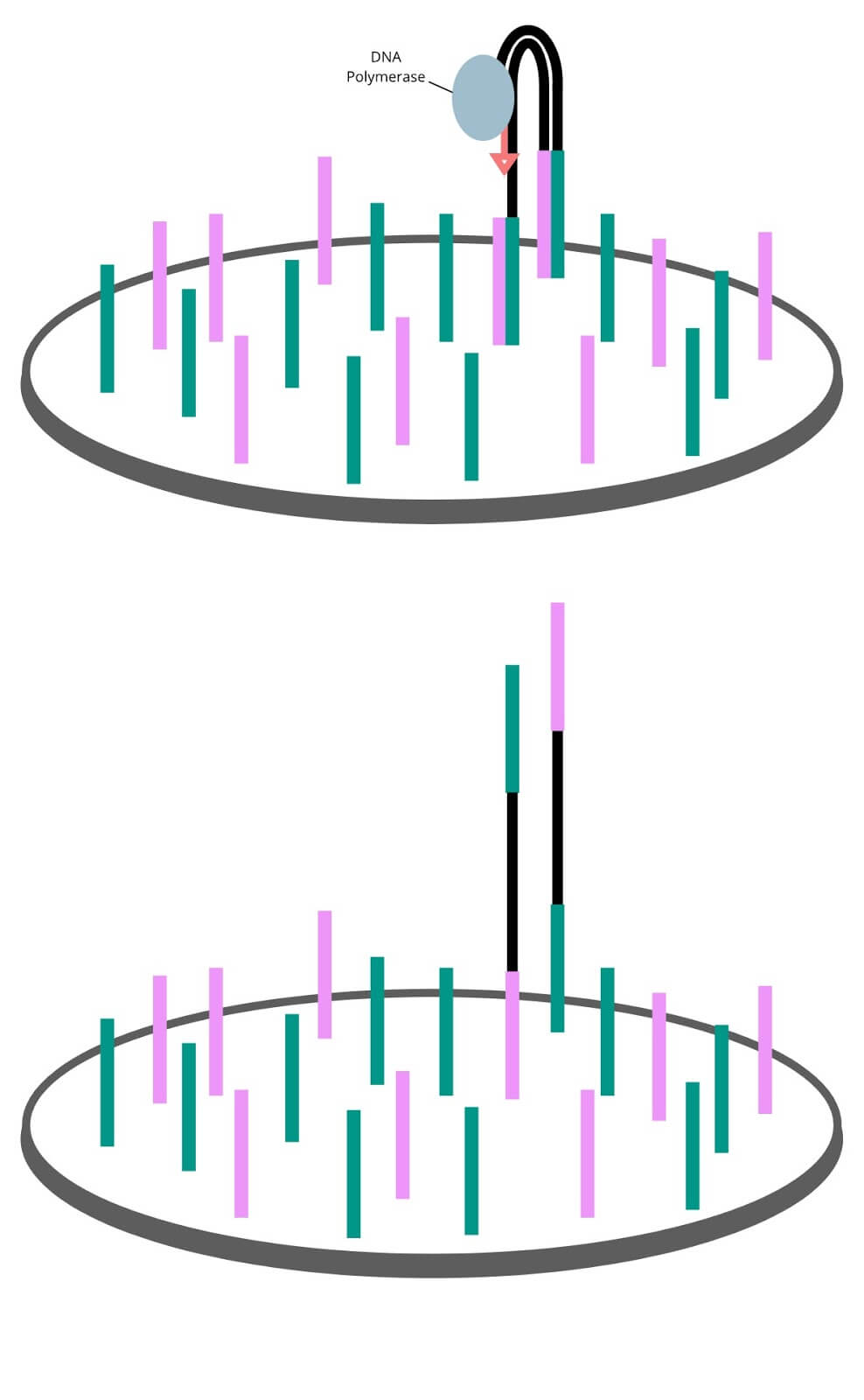
Sequencing by synthesis
Using enzymes and sequencing primers to kick-start the reaction, the forest of single stranded genomic fragments are filled in letter by letter. Each new nucleotide added is bound to a fluorescent marker and a terminator molecule that only permits a single molecule to be added at a time. Each of the 4 nucleotides have their own unique fluorescent marker color. The synthesis step proceeds in cycles in which letters are filled in, extra letters are washed away, colors are imaged, and terminator molecules are removed until the double stranded molecule is filled in.
The sequencing process runs in a cycle. The 4 fluorescently labelled DNA nucleotides (A, T, G, C) are flooded onto to the flow cell. Enzymes in the mixture will add this base onto the genomic strands whose first available letter is complementary to the added base. Unbound nucleotides are washed away, leaving just the incorporated fluorescent molecule, producing a clear fluorescent signal in each well that the sequencer will then image. The fluorescent label is cut off, and the cycle begins again with a different base. As new bases are added,Bridge amplification copies the single captured fragment into a forest of identical fragments. each well on the flow cell will change colors. By looking through the images, the sequence of color changes can be translated back into DNA sequences.

Once the sequencing run is finished, the sequencer compiles all of the spots from all of the images it takes, and converts it into millions of short sequences of DNA letters. These short reads are exported as a FASTQ file along with other details about the read quality, ready to be taken forward for data analysis.
Writers of the CoreGenomics Blog estimate that the HiSeq4000 can typically process 90 exomes in just 2 days. As Twist Bioscience’s Human Core Exome Kit is designed and synthesised with high uniformity, this number is pushed even higher.
Also, in the Exome Sequencing 101: Library Preparation blog we discussed a process called multiplexing. Multiplexing adds barcodes onto each patient’s genomic samples, which are also sequenced, allowing them to be pooled with other sequences containing unique barcodes. On one flow cell, our patient’s sample could be mixed with 100s of other patients’ genomic samples, all of which are sequenced simultaneously.
Here is the entire series:
Exome Sequencing 101: Part 1 – Library Preparation
Exome Sequencing 101: Part 2 – Target Enrichment
Exome Sequencing 101: Part 3 – Next-Generation Sequencing (NGS)
What did you think?
Like
Dislike
Love
Surprised
Interesting
